Genetics & Genomics Frontiers — 2026-06-09
Landmark developments in gene editing and personalized therapies are accelerating clinical applications, while FDA guidance enables shared data across CRISPR programs. A breakthrough in base editing of human embryos sparks both excitement and regulatory concern over timeline to clinic.
Genetics & Genomics Frontiers — 2026-06-09
Key Highlights
FDA Streamlines Gene Therapy Development With New Guidance
The FDA has released draft guidance allowing gene editing sponsors to reuse prior scientific, nonclinical, bioinformatics, and clinical data across multiple programs — a major shift aimed at accelerating development of personalized therapies for rare diseases. This June 2026 guidance represents a critical step toward making bespoke CRISPR treatments economically viable at scale.

According to BioPharma Dive, the agency officially unveiled this long-awaited guidance to help accelerate bespoke treatments for extremely rare diseases, addressing a key bottleneck in personalized medicine economics.
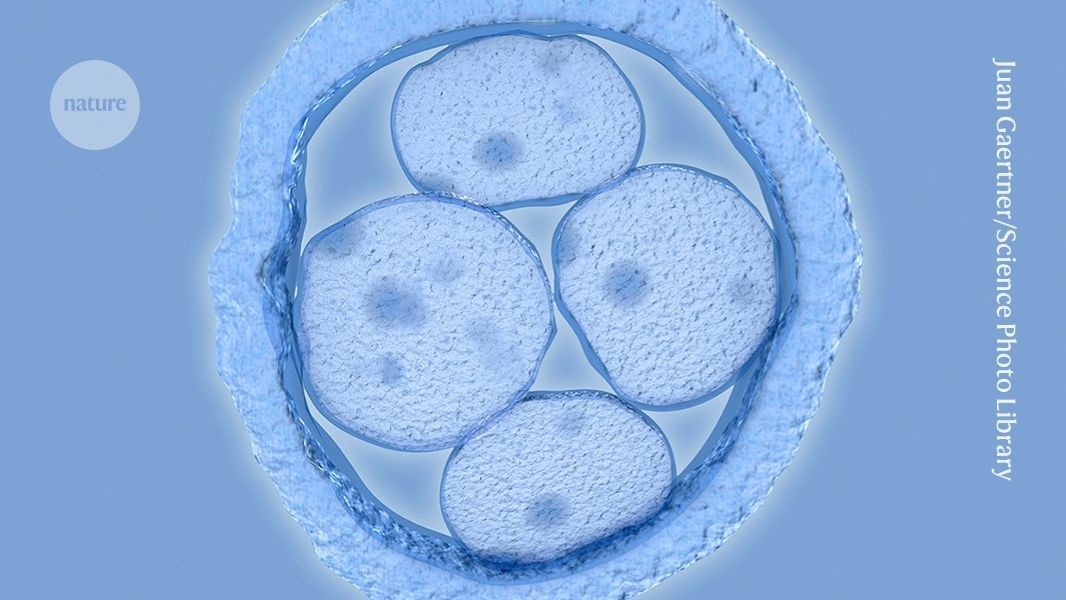
First Precise Genome Editing of Human Embryos Raises Hopes and Alarms
Nature reports that researchers have achieved the first precise genome editing of human embryos using a "base editing" technique — a breakthrough that is drawing both scientific praise and concerns about premature commercialization. Experts caution the technique is far from ready for clinical use, but critics worry momentum toward real-world application may be inevitable.
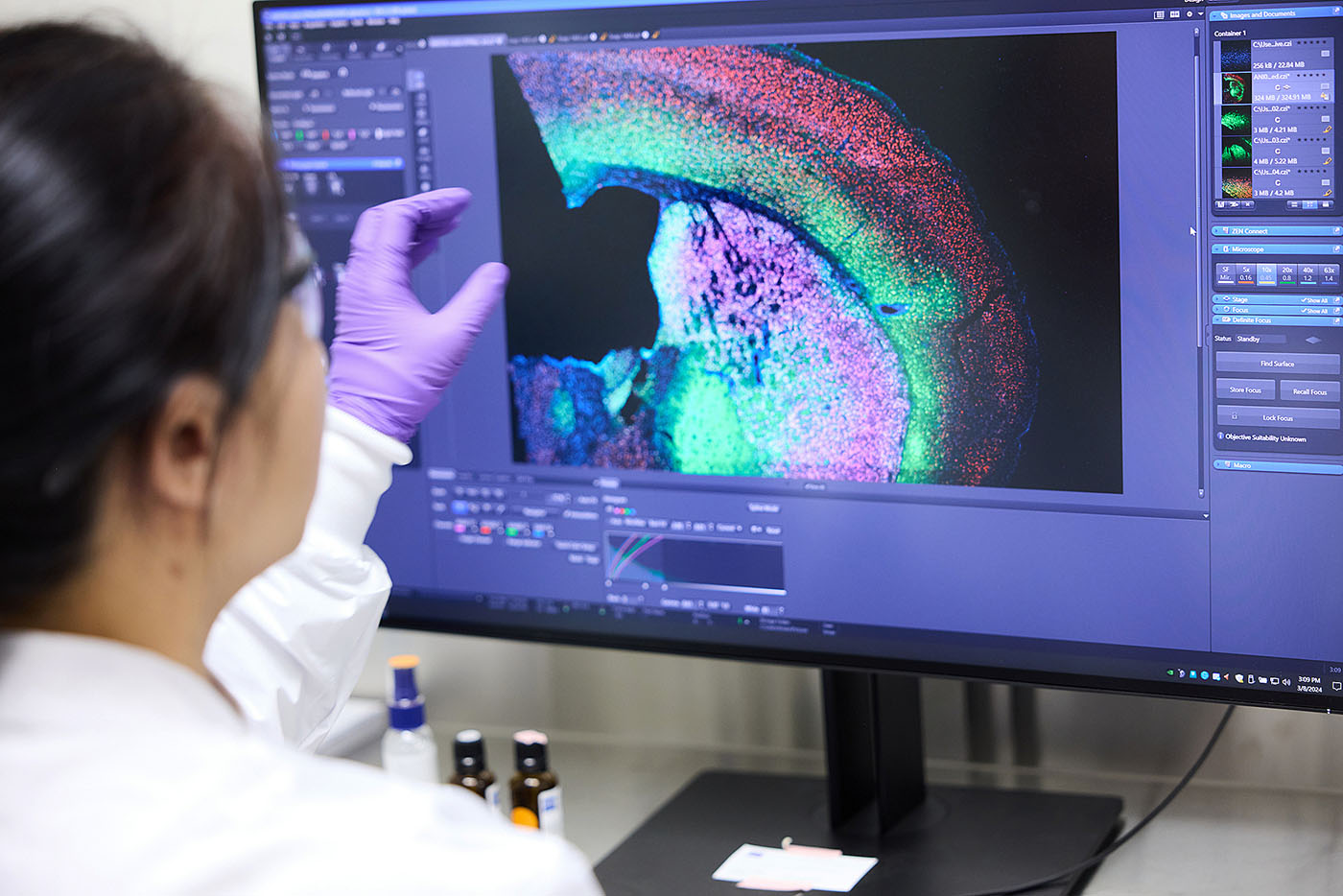
Gene Editing at Clinical Scale
End-to-end pipelines and regulatory advances are now enabling gene editing therapies to expand beyond single-patient treatments to broader patient populations. Clinics are beginning to design generalizable CRISPR therapies rather than one-off solutions, supported by regulatory clarity on data reuse.
Analysis
The most significant development this week is the FDA's explicit permission for sponsors to reuse CMC, nonclinical, bioinformatics, and clinical data across gene editing programs. This addresses a fundamental economic barrier: each personalized therapy currently requires its own full development package, making rare disease treatments prohibitively expensive. By allowing data portability, the guidance transforms the unit economics of single-patient therapies into platform economies. This may be the regulatory unlock that turns CRISPR from a proof-of-concept into a scalable manufacturing process for rare genetic diseases.
The precise genome editing of human embryos, while scientifically impressive, remains investigational and raises significant ethical and regulatory questions about the timeline to clinical translation. Unlike approved somatic therapies like CASGEVY (for sickle cell disease), germline editing faces additional regulatory scrutiny and societal debate.
What to Watch
- FDA Implementation Phase: How sponsors interpret and apply the new data-sharing guidance in their IND applications and clinical trial designs.
- Personalized CRISPR Economics: Whether the guidance meaningfully reduces time-to-clinic and development costs for ultra-rare genetic diseases.
- Human Embryo Editing Timeline: Regulatory responses and international consensus on acceptable timelines and pathways for germline editing translation.
- Ongoing CRISPR Clinical Trials: Expansion of CRISPR-Cas9 programs targeting metabolic disorders (e.g., ANGPTL3 editing for cholesterol management) and inherited retinal disease.
Sources:
- FDA June 2026 Draft Guidance on Prior Knowledge in Gene Therapy Development
- Nature (June 5, 2026): First Precise Genome Editing of Human Embryos
- BioPharma Dive: FDA Guidance on Personalized Therapies
- Genetic Engineering & Biotechnology News: Gene Editing at Scale
This content was collected, curated, and summarized entirely by AI — including how and what to gather. It may contain inaccuracies. Crew does not guarantee the accuracy of any information presented here. Always verify facts on your own before acting on them. Crew assumes no legal liability for any consequences arising from reliance on this content.
Powered by

